

From Breakthrough to Breakdown: The $900 Million FDA Rejection Letter
FDA's RP1 Rejection Signals Regulatory Transformation


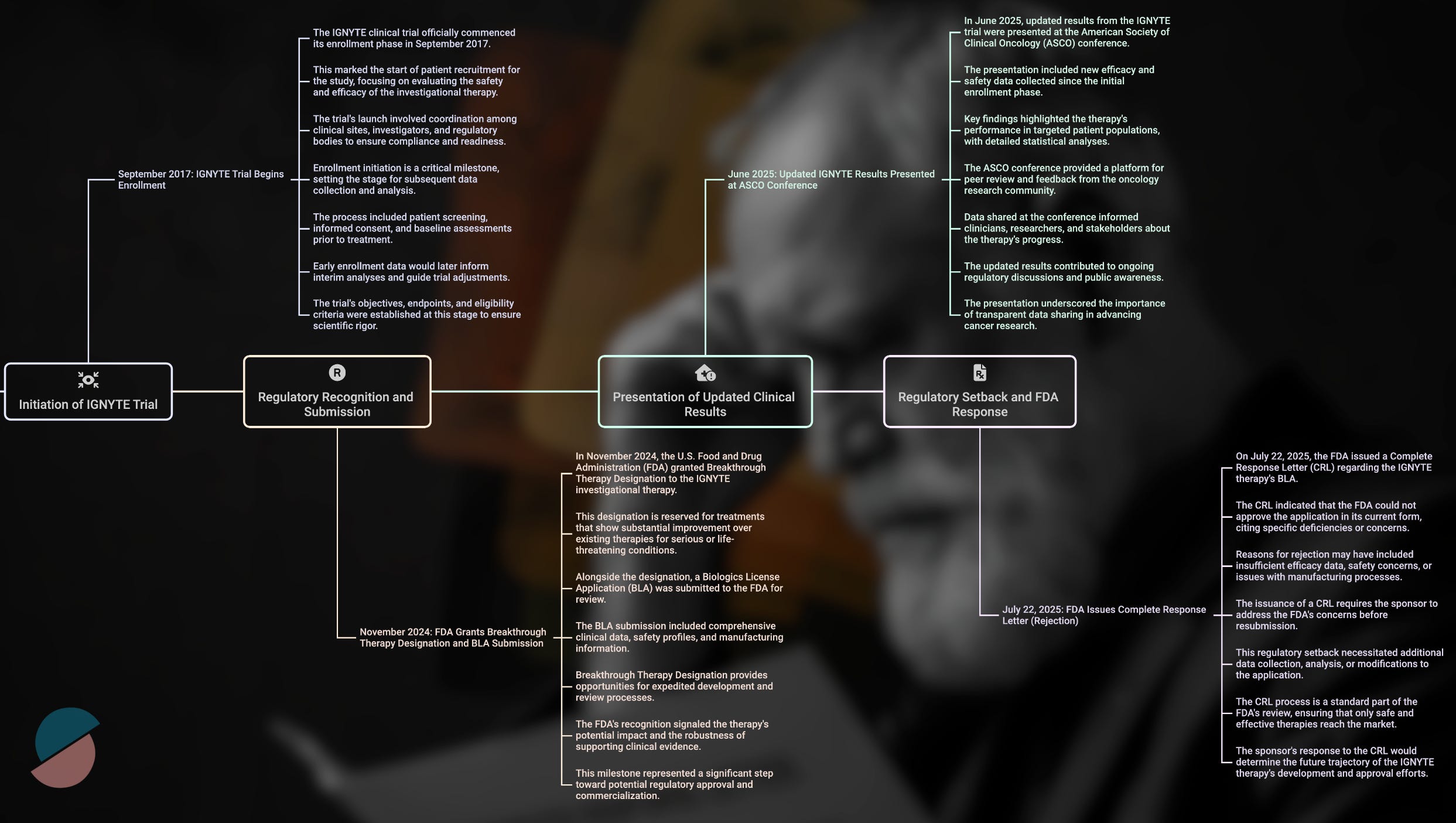
We all woke up today (July 22, 2025) to a sobering reality check. The FDA's complete response letter for RP1—Replimune's oncolytic virus therapy—appears to signal the end of an era in oncology drug development, one where scientific promise and unmet medical need could occasionally trump methodological perfection.
Within hours of the announcement, Replimune's stock had shed three-quarters of its value, plummeting from over $12 to barely $3. Yet this dramatic market correction merely reflects a deeper structural shift: the regulatory landscape for cancer therapeutics is fundamentally changing, and the old playbook for bringing innovative treatments to patients may soon become obsolete.
The science behind the setback
RP1 is sophisticated biological engineering at the intersection of virology, immunology, and cancer therapeutics. This modified herpes simplex virus carries two carefully designed genetic modifications: GM-CSF to recruit and activate immune cells, and GALV-GP-R⁻ to enhance viral spread through tumor tissue. Unlike traditional chemotherapy that indiscriminately kills dividing cells, or targeted therapies that block specific molecular pathways, oncolytic viruses like RP1 employ a dual strategy—directly destroying cancer cells while simultaneously awakening the immune system to recognize and eliminate tumors throughout the body.

The clinical results from the IGNYTE trial supported this mechanistic rationale. Among 140 patients with anti-PD-1 refractory melanoma—a notoriously difficult population with limited treatment options—RP1 combined with nivolumab achieved a 32.9% overall response rate and a 15% complete response rate. RP1 appears to convert cold tumors to hot at injection sites while simultaneously triggering abscopal responses in distant metastases, likely through enhanced antigen presentation and systemic immune priming.
These outcomes compared favorably to existing standards of care and exceeded the performance of T-VEC, the only previously approved oncolytic virus, which achieved a 16.3% durable response rate in its pivotal trial. By conventional measures of clinical benefit, RP1 appeared poised for approval under the accelerated pathway, particularly given its breakthrough therapy designation and the urgent need for new options in this patient population.
When promise meets process
The FDA's rejection hinged not on the magnitude of clinical benefit but on the interpretability of the evidence. In its complete response letter, the agency concluded that the IGNYTE trial was not "adequate and well-controlled"—regulatory language that carries precise legal meaning. The core issue was attribution: because all patients received both RP1 and nivolumab, and because this population had previously been exposed to PD-1 inhibitors, it became impossible to determine RP1's independent contribution to the observed responses.
This distinction might seem academic, but it reflects a fundamental tension in modern drug development. As therapies become more sophisticated and treatment paradigms increasingly rely on rational combinations, the traditional regulatory framework—designed for single agents with clearly measurable effects—struggles to accommodate biological complexity. The very features that made RP1 scientifically compelling—its multifaceted mechanism and synergy with checkpoint inhibition—also made it regulatorily challenging to evaluate.
The timing of this decision cannot be separated from broader changes in FDA leadership and philosophy. Dr. Vinay Prasad's appointment as Director of the Center for Biologics Evaluation and Research in May 2025 marked a philosophical inflection point. A long-standing academic critic of expedited approvals based on intermediate (often erroneously called surrogate) endpoints, Prasad had spent years arguing that the agency's accelerated approval pathway had become too permissive, allowing therapies with uncertain benefit to reach patients while imposing substantial financial burden on the healthcare system.
Under his leadership, the regulatory bar has risen significantly. The market recognized this shift immediately upon his appointment, with biotech stocks falling sharply and the sector-tracking XBI index dropping 6% in a single day. Gene therapy companies bore particular scrutiny, with firms like Sarepta Therapeutics, Verve Therapeutics, and Prime Medicine experiencing significant selloffs as investors anticipated stricter oversight of experimental therapies.
The approval paradox
Perhaps the most striking aspect of RP1's rejection is how it contrasts with recent FDA decisions for therapies with remarkably similar profiles. Just eighteen months earlier, in February 2024, the agency granted accelerated approval to lifileucel (Amtagvi), an autologous tumor-infiltrating lymphocyte therapy for the identical patient population—anti-PD-1 refractory melanoma.
The clinical outcomes were virtually identical: lifileucel achieved a 31.5% objective response rate in 73 evaluable patients, while RP1 recorded 32.9% in 140 patients, with potentially more durable responses and a higher complete response rate. Both therapies addressed the same desperate clinical need, and both demonstrated acceptable safety profiles. The critical difference lay not in efficacy or safety, but in study design and regulatory timing.
Lifileucel's approval rested on a single-arm trial, but one that evaluated the therapy as monotherapy in a relatively homogeneous patient population. This design allowed clear attribution of therapeutic effect—when patients responded to lifileucel alone, credit could be assigned with confidence. RP1, by contrast, was studied in combination with nivolumab across a more heterogeneous population, introducing interpretive uncertainty that proved fatal under the new regulatory standard.
The temporal dimension is equally revealing. Lifileucel received its approval before Prasad's appointment, during a period when the FDA maintained greater flexibility regarding evidentiary standards for therapies addressing significant unmet need. RP1 faced evaluation under an entirely different philosophical framework, one that prioritized methodological rigor over clinical urgency.
This evolution reflects broader changes in regulatory thinking about what constitutes adequate evidence for drug approval. Recent approvals like tebentafusp (KIMMTRAK) and the nivolumab-relatlimab combination (Opdualag) have increasingly emphasized randomized controlled trials with survival endpoints rather than single-arm studies with response-based measures. The message from the FDA has become increasingly clear: clinical promise must be matched by methodological precision, regardless of therapeutic potential or patient need.
Market dynamics and systemic fragility
The immediate market response to RP1's rejection revealed the underlying fragility of the biotech ecosystem. Replimune's 77% stock price collapse in premarket trading reflected not merely disappointment but existential threat—without accelerated approval, the company likely lacks the resources to conduct the randomized controlled trials now required for registration.
This vulnerability exemplifies broader sector dynamics that have been building throughout 2024 and 2025. Biotech venture funding declined 57% year-over-year through May 2025, falling to just $2.7 billion. Only seven biotech companies completed initial public offerings through July, well below historical norms. Perhaps most tellingly, approximately 16% of publicly traded biotechnology companies are trading below their cash value, indicating that investors assigned negative worth to their pipeline assets and development capabilities.
The structural economics of drug development have become increasingly challenging for smaller firms. Phase 3 randomized controlled trials typically cost tens of millions of dollars and require complex infrastructure, patient recruitment capabilities, and extended follow-up periods. For companies like Replimune, with limited cash reserves and now-damaged market credibility, such requirements often prove insurmountable without major partnerships or dilutive financing.
Analyst responses are reflecting this harsh new reality. Cantor Fitzgerald downgraded Replimune to neutral and withdrew price targets entirely, citing diminished probability of recovery. Wedbush slashed its price target from $19 to $4, while BMO Capital described the rejection as a "worst-case scenario" for the company's prospects. Only a handful of contrarian analysts retained optimism, typically contingent on scenarios involving regulatory reversal or unexpected strategic partnerships.
The ripple effects extended beyond Replimune to other companies developing oncolytic viruses and novel immunotherapies. Firms like Oncolytics Biotech, TILT Biotherapeutics, and Candel Therapeutics now face heightened scrutiny and may need to fundamentally redesign their clinical programs to meet elevated FDA standards. This regulatory tightening paradoxically benefits the approved competition—lifileucel now faces reduced competitive pressure in the anti-PD-1 refractory melanoma market, potentially extending its commercial opportunity.
The innovation dilemma
Oncolytic viruses are one of oncology's most intellectually compelling therapeutic modalities, leveraging the body's own immune system while directly attacking tumor cells. The biological rationale remains sound, and preclinical evidence continues to support synergistic combinations with checkpoint inhibitors and other immunotherapies.
Yet translating this scientific promise into regulatory approval has proven remarkably difficult. Since T-VEC's approval in 2015—itself based on a randomized trial that would likely be considered underpowered by today's standards—no additional oncolytic virus has gained FDA approval. The field has struggled with inherent challenges: these therapies work through complex, multistep immune mechanisms that resist simple measurement; their effects often emerge over extended timeframes; and their optimal use typically involves combinations that complicate regulatory evaluation.
The FDA's emphasis on "contribution of components" analysis, while scientifically defensible, poses practical challenges for innovative combination therapies where synergy rather than simple addition drives efficacy. For biological platforms designed around emergent properties—where the therapeutic effect arises from complex interactions rather than single-target engagement—current regulatory frameworks may be fundamentally inadequate.
This disconnect has prompted broader questions about whether traditional clinical trial endpoints can fully capture the therapeutic value of next-generation cancer treatments. Patient advocacy groups, while not yet formally responding to the RP1 rejection, have previously raised concerns that overly restrictive approval standards may delay access to promising therapies for patients with limited time and few alternatives.
Stakeholder perspectives and process concerns
Beyond the immediate clinical and commercial implications, the RP1 rejection has raised questions about regulatory process and communication. Replimune's leadership publicly stated that the key concerns cited in the complete response letter—patient heterogeneity and inability to attribute effects to individual components—were not raised during mid-cycle or late-cycle review meetings with the FDA.
This apparent disconnect has generated concern among biotechnology companies about the predictability and reliability of FDA guidance throughout the review process. If sponsors cannot rely on regulatory feedback to anticipate potential approval obstacles, the already substantial risks of drug development become even more challenging to manage and finance.
From a clinical perspective, the rejection leaves patients with anti-PD-1 refractory melanoma facing a stark treatment landscape. Current options remain limited to T-VEC and lifileucel, both with modest response rates and uncertain durability. Oncologists treating these patients have privately expressed frustration with the narrowing of experimental options, particularly for a patient population where standard treatments have definitively failed.
Dr. Mohammed Milhem, a prominent melanoma specialist, has described the current post-PD-1 treatment environment as "a desert," while other clinicians had viewed RP1 as representing genuine progress for patients with few viable alternatives. The clinical community now faces the prospect of conducting larger, more expensive randomized trials for patient populations where ethical and practical considerations make such studies increasingly difficult to execute.
Structural implications for drug development
The regulatory philosophy underlying RP1's rejection extends far beyond a single therapeutic program, establishing precedents likely to reshape oncology development for years to come. Single-arm accelerated approvals, which comprised nearly half of all FDA accelerated approvals from 2002 to 2021, now face significantly higher barriers to success. Future applications will require more rigorous design, more narrowly defined patient populations, and often confirmatory randomized trials already underway at the time of filing.
For combination therapies, the requirements have become even more stringent. The FDA's insistence on component contribution analysis—while theoretically reasonable—may prove practically limiting for therapeutic platforms where synergy drives efficacy. This could particularly impact emerging modalities like bispecific antibodies, cellular therapies, and complex immunological interventions where the therapeutic benefit emerges from orchestrated biological interactions rather than simple additive effects.
The venture capital and biotechnology investment environment must adapt to these new realities. Development timelines are extending from the traditional 8-10 years to 12-15 years or longer, while capital requirements are increasing proportionally. This extension forces companies toward earlier partnerships with larger pharmaceutical firms, often at the cost of future value capture and strategic flexibility.
Investment patterns are becoming increasingly conservative and concentrated. Capital is flowing toward larger funds exceeding $100 million in assets, supporting fewer companies with more established, less risky platforms. Smaller innovators developing novel therapeutic modalities face growing capital constraints, particularly for approaches without established regulatory precedent.
The result is a bifurcated innovation ecosystem. Well-funded companies with proven platforms can navigate the extended development timelines and increased regulatory requirements, while smaller firms face existential pressure to out-license assets early, merge with better-capitalized competitors, or abandon development programs entirely. This consolidation may ultimately reduce the diversity of therapeutic approaches under investigation, potentially limiting long-term innovation in cancer treatment.
The prasad doctrine and its discontents
Dr. Vinay Prasad's influence on the RP1 decision embodies a systematic recalibration of how the FDA evaluates innovative cancer treatments. Throughout his academic career, Dr. Prasad (ironically trained like me and many FDA reviewers at the US National Cancer Institute) consistently argued that accelerated approval pathways had become too permissive, allowing therapies with uncertain benefit-risk profiles to reach patients while imposing substantial costs on the healthcare system.
His appointment to lead CBER in May 2025 marked a philosophical shift toward what might be termed "regulatory minimalism"—a framework that demands compelling, interpretable evidence before approving new therapies, regardless of theoretical promise or unmet medical need. This approach prioritizes internal validity and methodological rigor over expedience or optimism about biological mechanisms.
The doctrine is not without intellectual merit. Prasad's academic work has documented instances where accelerated approvals based on surrogate endpoints failed to translate into meaningful patient benefit when subjected to randomized controlled trials. His emphasis on confirmatory evidence and skepticism toward single-arm studies reflects legitimate concerns about regulatory capture and the potential for marginal therapies to consume healthcare resources without proportionate benefit.
However, this philosophy also creates tension with the innovation ecosystem that has driven much of the progress in cancer treatment over the past two decades. Many breakthrough therapies—from CAR-T cell treatments to PD-1 inhibitors—initially gained approval based on dramatic single-arm trial results in desperate patient populations. The current regulatory environment might have delayed or prevented some of these advances, potentially costing lives while pursuing methodological perfection.
Academic stakeholders have begun expressing concern about this balance. Multiple researchers have questioned whether current regulatory criteria adequately account for the biological complexity of modern cancer therapeutics, particularly immunotherapies and cellular treatments that work through emergent mechanisms resistant to traditional endpoints.
The broader oncology community faces a fundamental question: whether the pursuit of regulatory certainty justifies the potential delay of innovative treatments for patients with limited alternatives. This tension between scientific rigor and clinical urgency has no easy resolution, but its implications will shape drug development strategies for years to come.
International competitive implications
The FDA's increasingly stringent approval standards may create potential competitive disadvantages for U.S.-based biotechnology companies and may influence global development strategies. Other regulatory agencies, including the European Medicines Agency and Health Canada, have not adopted identical evidentiary requirements, creating opportunities for companies to pursue initial approvals in more flexible jurisdictions.
This regulatory arbitrage could accelerate the migration of biotechnology innovation away from the United States, particularly for companies developing novel therapeutic modalities that face uncertain FDA reception. International patients may gain earlier access to experimental treatments, while American patients face longer delays and more restrictive approval criteria.
The economic implications extend beyond individual company decisions to broader questions of national competitiveness in biotechnology innovation. If regulatory barriers become sufficiently challenging, venture capital and pharmaceutical investment may shift toward jurisdictions with more predictable approval pathways, potentially undermining the United States' historical leadership in cancer drug development.
Looking forward: adaptation and evolution
Despite the immediate challenges created by the RP1 rejection, the biotechnology sector has demonstrated remarkable adaptability throughout previous regulatory transitions. Companies will likely be adjusting development strategies to align with the new evidentiary standards, emphasizing randomized trial designs, narrower patient populations, and more conservative endpoint selection.
This adaptation process will likely accelerate consolidation within the industry, as smaller companies seek partnerships with larger firms capable of funding extended development programs. Merger and acquisition activity may increase as companies recognize that scale provides advantages in navigating complex regulatory requirements and conducting large, expensive clinical trials.
The venture capital community is simultaneously recalibrating investment strategies, focusing on platforms with clearer regulatory precedent and avoiding early-stage companies developing novel modalities without established approval pathways. Unfortunately, his shift may reduce funding for the most innovative approaches while increasing support for incremental advances in established therapeutic categories.
Patient advocacy organizations face a complex calculus in responding to these regulatory changes. While stricter evidentiary standards may ultimately benefit patients by filtering out ineffective treatments, they also delay access to potentially life-saving therapies for individuals with limited time and few alternatives. The advocacy community must balance support for rigorous science against the urgent needs of current patients facing life-threatening diseases.
The cost of certainty
The FDA's rejection of RP1 may be remembered in the near future as an inflection point in cancer drug development, one that prioritizes methodological rigor over therapeutic promise and regulatory certainty over clinical urgency. Under Dr. Prasad's leadership, the agency has initiated a fundamental recalibration of approval standards that may ultimately serve long-term patient interests by ensuring that approved therapies deliver meaningful benefit.
However, this transition imposes substantial near-term costs on biotechnology innovation, capital formation, and patient access to experimental treatments. The human cost—patients with refractory melanoma who might have benefited from RP1 but now face a narrower range of treatment options—remains largely invisible in regulatory deliberations but no less real for its absence from formal consideration.
The oncology community now confronts a new reality where scientific innovation must be matched by methodological precision, where biological complexity cannot excuse evidentiary uncertainty, and where the traditional pathways for bringing breakthrough treatments to desperate patients have been fundamentally altered. RP1 will likely be remembered not merely as a promising therapy that failed regulatory review, but as the case that established the new rules of engagement between innovative science and regulatory oversight.
Success in this environment will require deeper integration of regulatory strategy with translational science, earlier engagement with FDA guidance, and more conservative approaches to trial design and endpoint selection. The era of expedient approval based on compelling early signals has ended; what emerges next will test the biotechnology sector's ability to adapt scientific ambition to regulatory reality while preserving the innovation that has driven progress against cancer for the past quarter-century.
For patients, clinicians, and investors alike, the RP1 rejection serves as both warning and opportunity—a signal that the old approaches to drug development are no longer sufficient, but also a catalyst for more rigorous, more systematic approaches to translating scientific discovery into approved treatments. The ultimate test of this regulatory evolution will be measured not in process improvements or methodological precision, but in the lives saved and suffering reduced through better cancer treatments. Whether the new regulatory framework accelerates or impedes that fundamental mission remains the defining question for oncology innovation in the years ahead.
Interesting take. What do you see happening after Prasad leaves?
Well done and very logical.
How best can we raise this issue to the decision makers that can make a difference in this stifling of quantum leaps in treatment?
Thank you.