

The Broken Instrument Beneath the Evidence
How the measurement substrate of oncology drug development systematically favors false negatives and why the field has not noticed

Every oncology drug approved based on tumor-based endpoints in the last twenty-five years rests on an instrument. The instrument is not a machine, not a molecule, not a laboratory assay. It is a rather arbitrary convention. A rule about how to press a ruler against a shadow on a screen and decide whether the shadow has grown. The convention is called RECIST (Response Evaluation Criteria in Solid Tumors) and it dictates, with the stubborn authority of orthodoxy, how major regulatory decisions in solid tumor oncology have been made since the late 1990s.
RECIST works like this: A radiologist examines a CT scan and selects up to five “target lesions,” the largest measurable tumors in a given organ. She measures each one along its longest diameter, sums those diameters, and compares the sum to a baseline. If the sum has shrunk by 30% or more, she writes partial response. If it has grown by 20% or more, she writes progressive disease. Anything in between is stable disease. Complete disappearance is a complete response. These four words, expanded into the binary of ORR (objective response rate: the fraction of patients who achieve complete or partial response), are what we submit to the FDA as evidence of efficacy. They are the basis on which drugs are approved, priced, and prescribed to patients with cancer.
The framework has the unusual property of being both ubiquitous and invisible. Few oncologists have heard of it. Almost no one in clinical practice actually uses it. It is the instrument by which drugs are certified, not the instrument by which they are administered. This is the first fact about RECIST worth holding onto. We will return to it.
I. Two readers, one scan
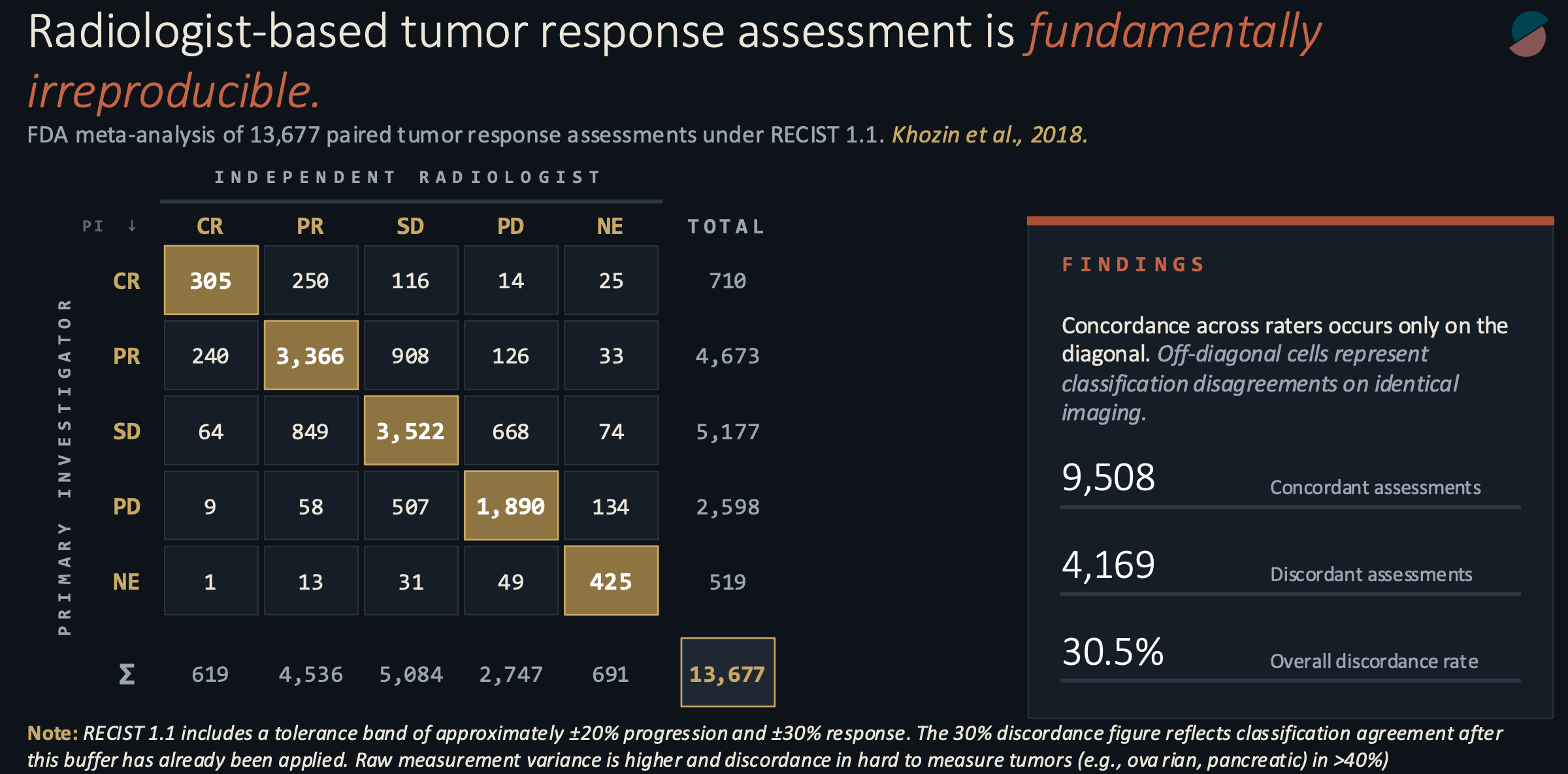
In 2017, while at the FDA, my colleagues and I pooled the paired radiology assessments from a decade of solid tumor registration trials. The structure of these trials has a quirk useful to our purpose: for regulatory submission, every scan is read independently by two qualified radiologists, one primary investigator review and the other a blinded radiologist at a central reading facility, each blinded to the other’s work. If they disagree, an adjudicator breaks the tie. The raw data, kept for internal auditing, records how often two expert readers, looking at identical images and applying identical criteria, reach identical conclusions.
Our pooled sample contained 13,677 paired assessments. The raw concordance rate was 69.5 percent. Two of every three paired reads agreed. The remaining third did not. In hard-to-measure malignancies such as pancreatic and ovarian cancers, the discordance exceeded 40% and, in some cases, reached the predictability of a coin flip.
This figure is higher than it ought to be. In the language of measurement theory, it corresponds to a weighted kappa somewhere between 0.30 and 0.55. Kappa is a statistic that asks how much two raters agree beyond what you would expect from chance alone; a value of 1.0 is perfect agreement, 0.0 is no better than coin flips. The number we observed sits at the lower boundary of what Landis and Koch, in their widely cited 1977 classification, called “moderate.” Moderate is what you would expect from a diagnostic tool in early development. It is not what you would expect from the instrument on which the entire oncology drug development enterprise rests.
The standard defense of the framework, when this figure is raised, is procedural. Discordance, the argument goes, is non-differential. It is distributed symmetrically across treatment and control arms. Therefore it does not invalidate the comparison. The p-value remains honest. This is partly true for randomized studies. It is also profoundly incomplete.
II. The attenuation
Here is what the procedural defense leaves out, beyond the fact that many cancer drugs are advanced in single-arm studies. When you inject random error into a comparison of two proportions, the observed difference between the proportions does not stay the same. It shrinks. This is a known result in epidemiology, tracing at least to the work of Irwin Bross in the 1950s, but it is rarely invoked in discussions of clinical trial design because most people prefer to believe that their measurements are not noisy.
The mathematics is merciful in its clarity. If the true response rate in the treatment arm is P(T) and the true response rate in the control arm is P(C), and each patient is misclassified with probability m, the observed difference between arms is the true difference multiplied by the factor (1 - 2m). At our measured discordance rate of thirty percent, that factor is 0.40.
A drug whose true advantage over standard of care is twenty percentage points of ORR, a substantial effect by any reasonable standard, appears in the trial record as a drug with an eight-point advantage. The biological signal has been attenuated by sixty percent before the statistician ever sees it.
Attenuation increases the sample size. Because the number of patients required to detect a given effect scales as the inverse square of that effect, a trial that would need two hundred patients to detect the true difference would need, under this measurement regime, approximately one thousand two hundred and fifty to detect the attenuated one. Most trials do not enroll 1,250 patients. Most trials are powered to detect the signal they were designed to detect, assuming the measurement faithfully transmits it.
The directional consequence is the one that matters, and it is rarely stated plainly. Random measurement error biases observed effects toward the null hypothesis. At the level of the measurement substrate, this produces a systematic excess of false negatives over false positives. A trial that fails to reach statistical significance is not necessarily a trial of an inactive drug. Some meaningful fraction of negative trials in solid tumor oncology are trials of active drugs evaluated with an instrument that could not see them.
No one knows the size of that fraction. The instrument, by its nature, erases the evidence of what it failed to detect.
III. Four boxes for a continuous world
The second property of RECIST is a property of encoding rather than of measurement. The underlying biological variable, the percent change in tumor burden over time, is continuous. It takes any value on the real line, bounded below by minus one hundred percent and above by whatever the tumor can manage. The framework transforms this continuous variable into an ordinal category with four levels, and then, for ORR, collapses those four levels into a binary. Something biologically rich becomes something statistically thin.
There is a way to quantify the loss, borrowed from information theory. A continuous percent-change measurement carries, under plausible distributional assumptions, between 5 and 8 bits of information per patient per scan. (A bit is the unit Claude Shannon introduced in 1948 to measure the content of a signal: one bit is the information gained from the outcome of a fair coin flip.) A four-level ordinal carries two bits, which is the base-2 logarithm of 4. A binary outcome carries one. The RECIST encoding, at its most generous, transmits at most a quarter of the information contained in the measurement it encodes. At ORR, it transmits less.
The effect is not merely a loss of resolution but a loss of biological distinguishability. An agent that produces a uniform 25% shrinkage in every treated patient, a remarkable result implying a real mechanism of action operating reliably across a population, can yield an ORR of zero. It is categorized, on the endpoint, as indistinguishable from placebo/no treatment. It is also indistinguishable, at the endpoint, from an agent that produces 19% growth. Three biologically distinct populations, trivially separable in the underlying distribution of percent change, collapse to the same point in the reported outcome.
This is the structural consequence of mapping a continuous variable through a step function. The step does what steps do. It discards what lies between its treads.
IV. Stochastic at the edge
The thresholds themselves introduce a third pathology, one that becomes vivid if you imagine the patient whose true response is not in the middle of a category but at its boundary.
For example, let’s look at a patient whose tumor burden has decreased by 28%. Two percentage points below the 30% partial response threshold. Stable disease by the rules, but barely. Suppose the inter-reader standard deviation on the sum of diameters, consistent with the discordance data, is about ten percentage points. The probability that a given reader assigns this patient to the PR category, under a normal approximation, is the probability that the reader’s estimate falls below -30%, which works out to about 42%.
Forty-two percent. The same biology, the same scan, the same criteria, yields partial response in forty-two of one hundred reads and stable disease in fifty-eight. The categorical outcome is, at the boundary, approximately a coin flip. And the reported trial effect depends on how many of these coin flips land on the side that counts.
This threshold instability does more than add variance. It creates a selection pressure that propagates backward into the development pipeline. Because ORR binarizes sharply at minus thirty percent, a sponsor who wishes to maximize the probability of clearing that threshold is rewarded for advancing agents capable of producing deep responses in a subset of patients. Agents that produce shallow but consistent cytoreduction across a population, even when the underlying biology is meaningful, are structurally disfavored. They cannot clear the threshold often enough to generate an ORR that the machinery can see. And our machinery is impatient. When meaningful tumor responses are not seen in early clinical trials, the entire program is at risk.
The shape of the modern oncology pipeline, its preferential enrichment for drugs that drive responses visible to the human eye, is a direct consequence of our endpoints. We have been selecting not for what can prolong survival or increase quality of life, but for what our instruments can detect.
V. The instrument that does not exist outside the trial
The third property is the most easily stated and the most commonly overlooked. RECIST is a rather artificial clinical trial instrument. It is not a clinical tool. Community oncologists, the people who actually treat the overwhelming majority of patients with cancer, do not compute sums of diameters. They do not segment target lesions into protocol-defined categories. They read narrative radiology reports written in ordinary English (”stable appearance of the right upper lobe mass”) and make decisions on the basis of clinical judgment integrated with that narrative.
Which is to say: the endpoint that certifies a drug’s efficacy in the trial setting is not the endpoint that governs its use in the practice setting. The variable on which approval rests is not present in the data that routine care generates. This is the oncology equivalent of approving, say, statins on the basis of LDL reduction only to find out that community physicians use blood pressure to administer and titrate therapy.
When we attempt to validate oncology clinical trial findings against real-world evidence, using electronic health records and community radiology reports, we cannot reconstruct a RECIST assessment. The comparison has to be made at a different level of abstraction, using different variables, and the correspondence between the two settings, at the level of the individual patient or the individual drug, remains structurally unverifiable.
The conventional response to this observation is that RECIST is a surrogate for clinical benefit rather than the benefit itself. A surrogate is a measurable variable used as a proxy for the outcome of real interest, which in oncology is almost always overall survival. The real question, on this view, is whether RECIST-based endpoints predict overall survival. The meta-analytic answer, accumulated across hundreds of trials and now reasonably mature, is that they do so imperfectly. PFS-to-OS correlations in solid tumor oncology routinely yield coefficients of determination below 0.5. ORR-to-OS correlations are lower. The surrogate explains less than half of the variance in the outcome it is supposed to predict.
The framework persists, then, less because of its predictive validity than because of its institutional entrenchment. It is the thing we all agreed to use. Agreement has its own gravitational pull, and for a long time the conditions that would permit disagreement did not exist.
VI. What the endpoint could not see
An instrument with 30% reader discordance, indicating moderate agreement by standard statistical classification, can attenuate observed effects by 60% and inflate required sample sizes sixfold. The attenuated signal is then projected onto an ordinal encoding that transmits at most two bits per scan and produces stochastic categorization at the thresholds where the reported effect is determined. The resulting endpoint is computable only within the trial setting and cannot be reconstructed from the data generated by clinical practice after approval.
The directional bias of the whole apparatus is toward false negatives. It favors drugs whose mechanism produces a small number of responses that are deep enough to be detected by the human eye over drugs whose mechanism produces a large number of modest responses, but can prolong survival or improve quality of life. It cannot see agents whose signals fall below the threshold for categorical instability, and it cannot distinguish, at the endpoint, between agents that differ by biologically meaningful amounts.
The statistical machinery layered on top of this substrate, all the careful p-value thresholds and hierarchical testing procedures and alpha-spending rules, is bounded by what the substrate permits. No refinement of the hypothesis test can recover information that the measurement did not transmit.
The cost is not just borne by the sponsors whose trials failed. It is borne by the patients whose therapies were never developed because the endpoint could not see them.
VII. The substrate is replaceable now
The conditions that produced RECIST in the late 1990s are no longer operative. Volumetric segmentation of tumors, computationally prohibitive a few years ago, is now a solved problem. Foundation models for medical imaging, trained on orders of magnitude more data than any individual radiologist will read in a career, are mature. Automated assessment at scale, across both trial and community settings, is a deployed capability.
Over the past several years, I have been working on a replacement substrate at Project Data Sphere. We call it the Total Tumor Burden Index. It is volumetric rather than diametric, which reduces reader-dependent variance. It is continuous rather than categorical, which eliminates both the information loss of ordinal encoding and the instability at categorical boundaries. It is machine-generated rather than reader-dependent, which removes the dominant contemporary source of measurement noise and allows deterministic reproducibility across sites and across re-reads. And it is equally deployable in trial and community settings, which means the trial measurement and the practice measurement can, for the first time, be the same variable.
The regulatory path will take time. Prospective comparison with RECIST in contemporary datasets will take time. Integration across imaging infrastructure will take time. None of these barriers is technical. The technical conditions for the replacement are being satisfied with increasing precision.
VIII. Coda

The measurement substrate in solid-tumor drug development is not neutral. It has been shaping the pipeline for twenty-five years in ways the field has not made fully explicit to itself. The attention has remained on the superstructure, the p-values, the statistical analysis plans, and the regulatory pathways, when the leverage is actually in the layer underneath. Replacing the measurement, rather than refining the statistics computed upon it, is the tractable path forward. The conditions for that replacement are, at last, in place.
What we measure determines what we can find. What we can find determines what we develop. What we develop determines who is treated. The ruler we have been pressing against the tumor for a generation is not the only ruler available. It is simply the one we inherited. And that inheritance is due for review.
Important to mention most modern oncology trials are incorporating ctDNA and pathologic response. Surrogates and biomarkers even RECIST aren’t perfect by any means but in real life they do correlate well with disease.