

42 Seconds
On daraxonrasib, the pocket that had to be invented, and what the standing ovation at ASCO really means


I. Chicago, May 31
Like many of my colleagues, I was in the room for the phase 3 results of RASolute 302, a randomized trial testing daraxonrasib, a first-in-class oral inhibitor of active RAS, against standard chemotherapy in patients with previously treated metastatic pancreatic cancer.
The presentation was a late-breaking plenary at ASCO’s 2026 annual meeting at McCormick Place in Chicago. When the Kaplan-Meier curves appeared on the screen, the two survival lines pulling apart with an unusually wide and stable separation, the audience stood and applauded before Brian Wolpin, director of the Hale Family Center for Pancreatic Cancer Research at Dana-Farber and lead investigator of the trial, had finished speaking.
It lasted 42 seconds.
When the applause subsided, Wolpin noted that the time had not been built into his talk.
These results deserve to be understood on their clinical merits, which are substantial. But they also deserve to be understood as the product of a particular kind of scientific thinking, one that required forty years of accumulated failure before we could ask the right question.
II. The disease that has resisted almost everything
Pancreatic ductal adenocarcinoma kills most of the people it touches. In 2026, roughly 67,500 Americans will be diagnosed, more than half with metastatic disease at presentation. For those patients, the five-year survival rate is around 3%. When first-line chemotherapy stops working (and it almost always does), second-line treatment has historically offered a median overall survival of 6 to 7 months and a median progression-free survival of 3 to 4 months, at the cost of substantial toxicity.
One of the central molecular drivers has been known for decades. Oncogenic RAS mutations are present in more than 90% of pancreatic tumors, most involving substitutions at KRAS codon 12. These mutations lock RAS in a GTP-bound active state, driving proliferative signaling the cell cannot turn off. The therapeutic conclusion has always seemed obvious: inhibit active RAS, and you could reach nearly every patient.
For forty years, nobody could.
III. The geometry of failure
Gilbert Ryle, the British philosopher, introduced the concept of a category mistake in 1949 to describe what happens when someone searches for a thing in a place where, by the nature of the category being used, it cannot exist. His canonical example is a visitor touring Oxford who walks past every college, every library, every building, and then asks: but where is the University? She had seen the University. She failed to recognize it because she was looking for a different kind of thing. The error was conceptual before it was empirical.
The forty-year failure to inhibit RAS-GTP was, at its root, a category mistake of this kind.
Drug discovery has long operated within what is known as a substance ontology: a framework in which the fundamental objects of interest are independent entities that bear properties. Proteins have pockets. Targets have binding sites. A protein is druggable or it is not. RAS-GTP presented exactly the kind of problem this framework was least equipped to solve: a smooth, largely featureless surface, no deep binding pocket, and picomolar affinity for its natural substrate GTP, making competitive displacement essentially impossible. We treated druggability as an intrinsic property of the protein itself, found no such property in active RAS, and concluded it could not be done. So we looked downstream.
In 2013, Kevan Shokat, professor of cellular and molecular pharmacology at UCSF and a co-founder of Revolution Medicines, cracked this open by identifying a transient pocket on the KRAS G12C mutant. That insight led to sotorasib and adagrasib, validated RAS as a pharmacological target, and changed the conversation. But these drugs bind KRAS in its GDP-bound inactive conformation; they reach RAS only when it is already off. And G12C is rare in pancreatic cancer. For the patients who most needed RAS inhibition, the first generation was, by design, largely beside the point.
The central question remained: how do you inhibit active RAS, GTP-bound and constitutively firing?
IV. What nature knew, and what we failed to generalize
The breakthrough that produced daraxonrasib did not come from finding a hidden pocket on RAS. It came from escaping the assumption that druggability had to be a property of RAS itself. The decisive question was relational: what if the drug did not have to bind RAS alone?
This required abandoning an assumption so deeply embedded in traditional drug discovery that most of us had stopped noticing it as an assumption. One target, one protein, one binding site. The paradigm is powerful and ubiquitous. For active RAS, it was a dead end.
What is striking in retrospect is that nature had been demonstrating a different paradigm for decades, and we had been watching. Rapamycin does not inhibit mTOR by occupying a pre-existing mTOR pocket; it first binds FKBP12, and the resulting complex engages mTOR through an interface neither partner could access alone. Cyclosporin A recruits cyclophilin A to inhibit calcineurin. Thalidomide forces cereblon to bind and degrade transcription factors it would otherwise ignore. These are among the most consequential drugs in medicine, and they share a common logic: a small molecule conscripts an endogenous protein to create a new interaction that blocks a biological process indirectly. Before active RAS could be drugged, nature had already shown the way.
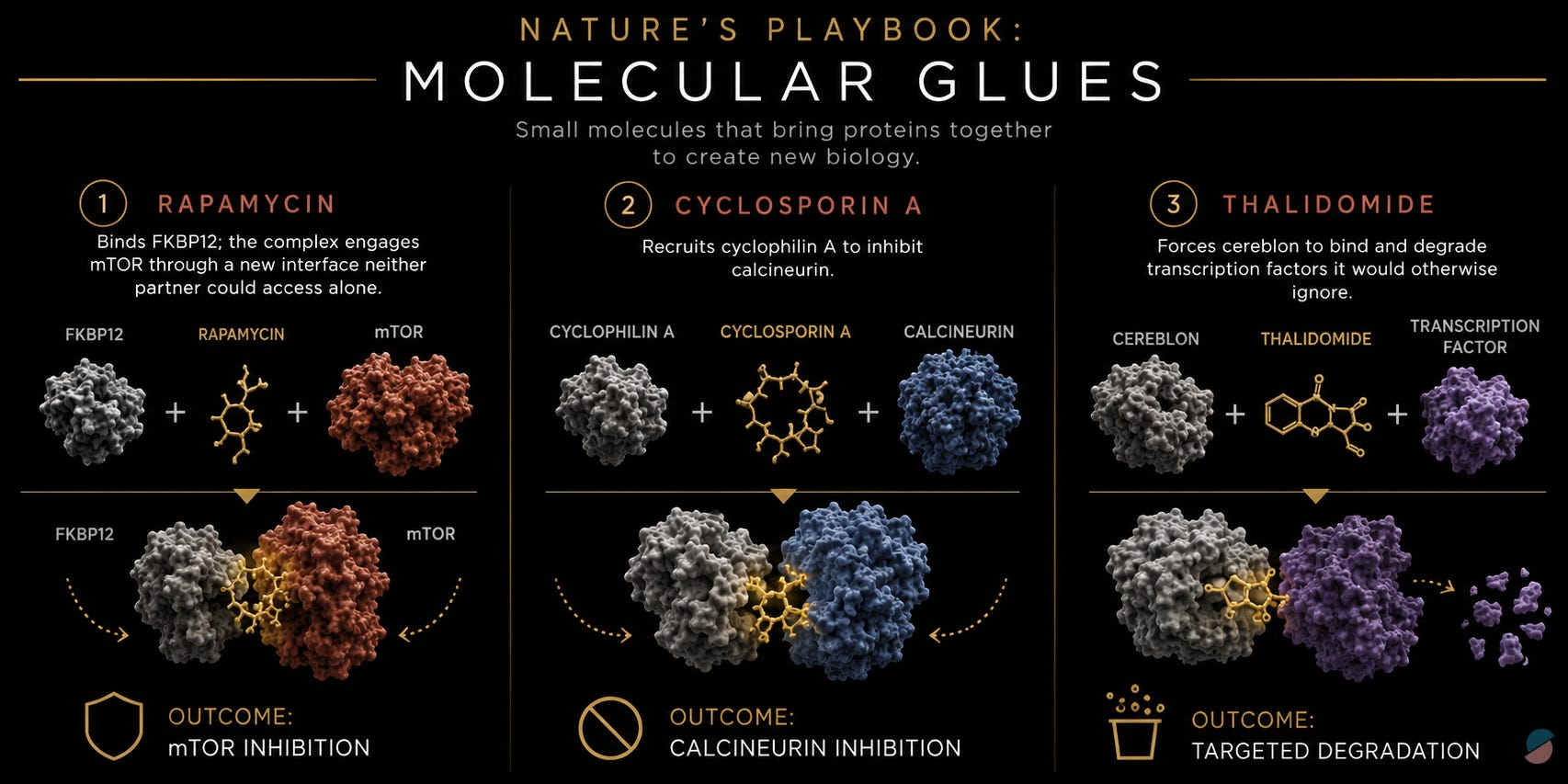
But none of these drugs emerged from a rational design program. Rapamycin is a natural product isolated from a soil bacterium found on Easter Island in 1972. Cyclosporin A came from a Norwegian fungus identified in a Sandoz screening program. Thalidomide was a sedative whose mechanism in cancer was reverse-engineered decades after it was withdrawn from the market for causing birth defects. In each case the sequence was the same: discover the molecule, observe what it does, spend years understanding why. These drugs did not begin as a design principle. They ended up revealing one, and we largely filed the lesson away. Each mechanism was admired as elegant and categorized under its own domain (immunophilin pharmacology, cereblon biology) without asking whether the underlying logic could be deliberately applied to problems that had resisted direct inhibition.
This is a recognizable pattern in science. Paradigm-shifting evidence accumulates in plain sight for years before someone asks whether it generalizes. The data were there. The inference was not.
Shokat’s group made that inference in 2019, publishing proof-of-concept work showing that bifunctional ligands could recruit cyclophilin A to GTP-bound RAS and block its interaction with B-Raf. This was not yet a drug. But it was the answer to a question we had not been asking.
V. Sanglifehrin A: the handle that made it possible
Revolution Medicines, founded in 2014 by Martin Burke, a chemist and natural product synthesis pioneer at the University of Illinois, Michael Fischbach, a microbiologist and biosynthesis expert at Stanford, and Shokat himself, had an explicit founding thesis: natural products could be reverse-engineered into precision medicines for targets that conventional drug discovery could not reach. The company now had the concept. It needed the chemistry.
The scaffold came from a natural product discovered by Novartis in 1999 and never developed as a drug: sanglifehrin A, a bacterial macrocycle with high affinity for cyclophilin A. Its value was structural. Its binding mode leaves a solvent-exposed face pointing outward, a chemical vector free to be linked to a RAS-binding pharmacophore. One face buried in cyclophilin A, the other engineered toward RAS-GTP.
The team built sanglifehrin-inspired bifunctional molecules and solved the crystal structures of the resulting tricomplexes with KRASG12C. What those structures revealed could not have been predicted from studying either protein in isolation. The drug sits in a composite pocket that is partly cyclophilin A and partly RAS: a neomorphic interface that does not exist when either protein is present without the other. The molecule remodels cyclophilin A’s surface to create a high-affinity interface with active KRASG12C. The pocket was not discovered. It was created.
VI. From G12C to every RAS: the making of daraxonrasib
The first clinical molecules from this platform, elironrasib for G12C and zoldonrasib for G12D, were mutation-selective, exploiting covalent chemistry anchored to specific mutant residues. Elegant, but insufficient for pancreatic cancer, where the full spectrum of oncogenic RAS variants collectively accounts for nearly the entire patient population.
Daraxonrasib was built around a broader premise: engineer a molecule that engages the conserved switch regions shared across all oncogenic RAS variants rather than the residues that distinguish one mutation from another. The result is a reversible, noncovalent molecular glue: macrocyclic, orally bioavailable, mutation-agnostic.
The mechanism is worth holding precisely. Daraxonrasib binds cyclophilin A first, forming a binary complex that then engages RAS-GTP across the composite interface. The tricomplex blocks downstream effectors (RAF, PI3K) from reaching the RAS surface. The drug does not fit into RAS. It fits into a relationship between two proteins, a relationship it calls into being. For G12X mutants there is an additional effect: daraxonrasib can stimulate GTP hydrolysis, actively accelerating RAS back toward its inactive state rather than merely blocking its downstream signals.
That distinction matters. Daraxonrasib is not a better RAS inhibitor in the conventional sense. It is a different way of thinking about what an inhibitor can be.
VII. What 13.2 months means
RASolute 302 enrolled 500 patients across 59 sites in six countries, randomized 1:1 between daraxonrasib 300 mg once daily and investigator’s choice of chemotherapy. Results were published in the New England Journal of Medicine the same day they were presented at ASCO.
Of enrolled patients, 91.8% had RAS G12 mutations, the primary endpoint population. In that population, median overall survival was 13.2 months with daraxonrasib versus 6.6 months with chemotherapy, a hazard ratio of 0.40 representing a 60% reduction in the risk of death. At 12 months, 53.3% of daraxonrasib patients were alive versus 18.7% on chemotherapy. Median PFS was 7.3 versus 3.5 months. Objective response rate was 33.2% versus 11.8%.
The context that makes 13.2 months remarkable: the landmark first-line regimens in this disease (FOLFIRINOX, gemcitabine plus nab-paclitaxel, NALIRIFOX) achieved median overall survival of 8.5 to 11.1 months in previously untreated patients. Daraxonrasib, in patients who had already exhausted those regimens, compares favorably with all of them.
Beyond survival, time to pain deterioration was 9.0 versus 3.7 months. Patients were not only living longer. They were feeling better longer. Treatment-related discontinuation occurred in 1.2% of daraxonrasib patients versus 11.2% on chemotherapy. Hematologic toxicities and peripheral neuropathy, the defining burdens of chemotherapy in this disease, were largely absent.
FDA expanded access was authorized in May 2026. The broader question remains: how quickly can a system built around sequential evidence review deliver a result this large to the patients who need it?
VIII. A question worth sitting with
There is something in this story worth dissecting.
When Wolpin showed those survival curves at ASCO, the knowledge that daraxonrasib halves mortality in metastatic pancreatic cancer became public. But that knowledge existed at the data cutoff in February. It existed, in effect, from the moment the trial was powered enough to see it. And yet the patients sitting in oncology clinics that same week, the ones not enrolled in RASolute 302 and not navigating expanded access, were receiving second-line chemotherapy with a median survival of under seven months, because the drug that could double that had not yet cleared the regulatory process required to reach them.
This is the gap we need to talk more about. Not a failure of any single institution, not a problem unique to daraxonrasib, but a structural feature of how we move evidence from signal to treatment. The trial completed enrollment in November 2025. The data cutoff was February 2026. Results were presented publicly in June. Submission, review, approval, and prescribing infrastructure still lie ahead. Each step is defensible in isolation. Together, they add up to months; and in pancreatic cancer, months are the unit of survival.
The architecture we built to evaluate evidence was designed for an era of slower science, slower information, and limited computational capacity. It made sense then. The question worth asking now is whether a system built around collecting data, freezing it, submitting it, and reviewing it as a completed artifact is still the right model when the signal is a hazard ratio of 0.40 on a pre-specified primary endpoint in a 500-patient randomized trial, and the disease being treated measures survival in single-digit months.
IX. The regulatory paradigm needs its own conceptual leap
The daraxonrasib story is about changing the unit of analysis. We could not inhibit active RAS because we were looking for a single-protein binding site that did not exist. The breakthrough came when someone changed the question from “where is the pocket?” to “what relationship could we create?” The same logic applies to how we move evidence from trial to patient.
The current model treats drug evaluation as a sequential, bounded process: conduct the trial, freeze the data, submit a dossier, review it, decide. I called this the HMS Salisbury model in an earlier essay, “The ship and the signal”: the idea that medical evidence must be collected, transported, and unpacked before it can inform a decision, the way James Lind’s citrus observations had to cross the Atlantic before anyone acted on them. It was the right architecture for its time. It is increasingly misaligned with a world of molecularly targeted therapies, large and unambiguous signals, and diseases that measure survival in single-digit months.
The FDA’s first real-time clinical trial pilot (AstraZeneca’s TRAVERSE study in mantle cell lymphoma and Amgen’s STREAM-SCLC study in limited-stage small cell lung cancer) proves the model can change. In that framework, endpoint and safety signals reach reviewers as they emerge rather than arriving frozen in a dossier after trial completion. It is not yet a transformation. It is a proof of concept, and a sign that a fundamentally different paradigm is within reach.

X. What the standing ovation was really for
I have been to many ASCO plenary sessions. I have not seen many moments like the one on Sunday.
I think people standing in that hall were not applauding a hazard ratio. They were applauding the end of a forty-year impasse in a disease where failure had become the baseline expectation, and where the same grim prognosis had been delivered to patients year after year despite everything the field had tried.
But the full measure of what Sunday means depends on what we do next.
The scientific paradigm changed when someone asked a different question about where a binding site could be found. The regulatory paradigm needs the same move: away from evidence as a frozen artifact reviewed after the fact, toward evidence as a continuous signal evaluated in real time. The science and the system have to advance together, or the science advances into a bottleneck.
It took forty years of failure, a philosopher’s insight about category mistakes, three natural products that nobody designed, a bacterial macrocycle that sat unused for twenty-five years, and a company founded on the belief that nature encodes answers to questions we have not yet learned to ask. The regulatory infrastructure capable of delivering the resulting therapy to patients without avoidable delay does not fully exist yet either.
Creating it will require the same thing that created daraxonrasib: the willingness to question whether the current frame is the right one.
The question changed. That is why the room stood up.
Now the room has to go to work.
Not to mention that the vision needs to be OS for YEARS not months, and the "evidence" starts with One and builds on similar phenotypes. The "system" needs to drop its obsession with "randomized" trials for huge cohorts of patients with differing biological narratives (of both host and invader).